GDC MAF Format v.1.0.0
Introduction
Mutation Annotation Format (MAF) is a tab-delimited text file with aggregated mutation information from VCF Files and are generated on a project-level. MAF files are produced through the Somatic Aggregation Workflow. The GDC produces MAF files at two permission levels: protected and somatic (or open-access). One MAF file is produced per variant calling pipeline per GDC project. MAFs are produced by aggregating the GDC annotated VCF files generated from one pipeline for one project.
Annotated VCF files often have variants reported on multiple transcripts whereas the MAF files generated from the VCFs (*protected.maf) only report the most critically affected one. Somatic MAFs (*somatic.maf), which are also known as Masked Somatic Mutation files, are further processed to remove lower quality and potential germline variants. For tumor samples that contain variants from multiple combinations of tumor-normal aliquot pairs, only one pair is selected in the Somatic MAF based on their sample type. Somatic MAFs are publicly available and can be freely distributed within the boundaries of the GDC Data Access Policies.
The GDC MAF file format is based on the TCGA Mutation Annotation Format specifications, with additional columns included.
Note: The criteria for allowing mutations into open-access are purposefully implemented to overcompensate and filter out germline variants. If omission of true-positive somatic mutations is a concern, the GDC recommends using protected MAFs.
Somatic MAF File Generation
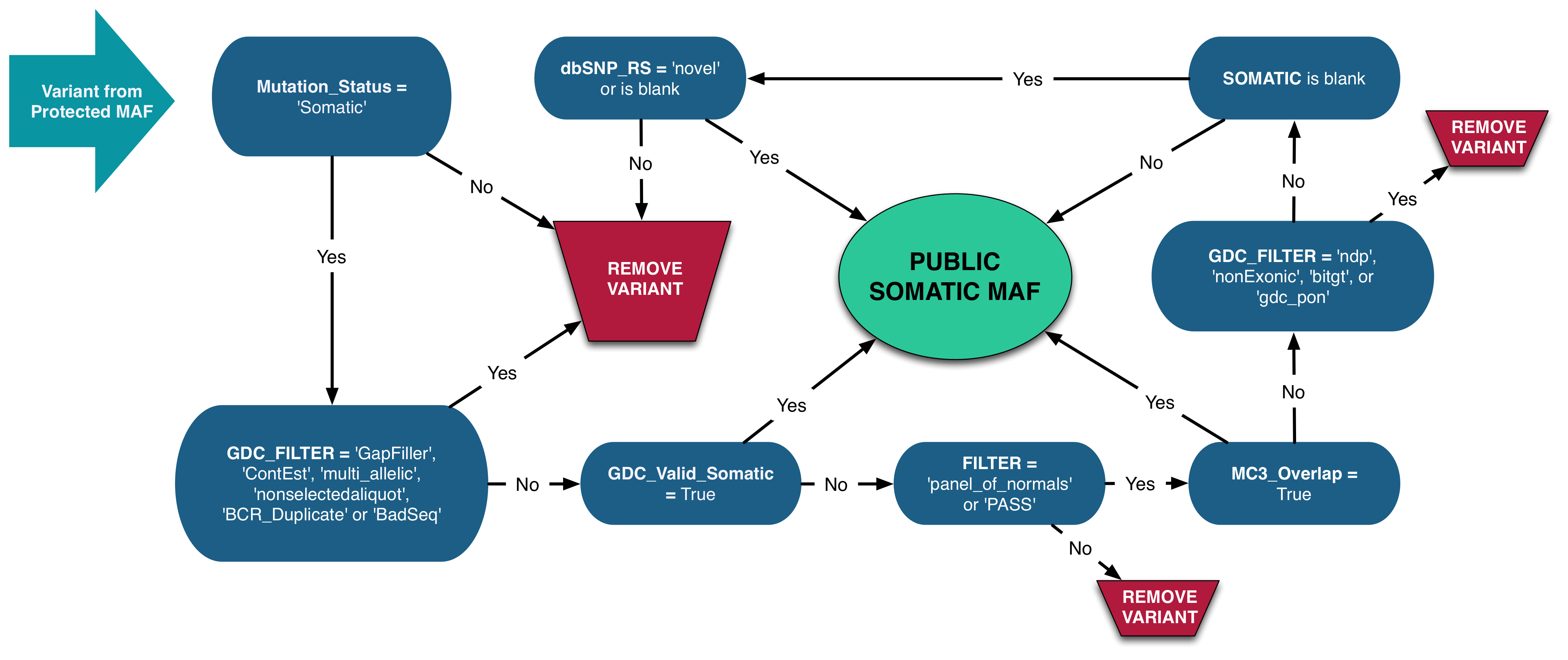
The process for modifying a protected MAF into a somatic MAF is as follows:
- Aliquot Selection: only one tumor-normal pair are selected for each tumor sample based on the plate number, sample type, analyte type and other features extracted from tumor TCGA aliquot barcode.
- Low quality variant filtering and germline masking:
- Variants with
Mutation_Status != 'Somatic'orGDC_FILTER = 'Gapfiller','ContEst','multiallelic','nonselectedaliquot','BCR_Duplicate'or'BadSeq'are removed. - Remaining variants with
GDC_Valid_Somatic = Trueare included in the Somatic MAF. - Remaining variants with
FILTER != 'panel_of_normals'orPASSare removed. Note that theFILTER != panel_of_normalsvalue is only relevant for the variants generated from the MuTect2 pipeline. - Remaining variants with
MC3_Overlap = Trueare included in the Somatic MAF. - Remaining variants with
GDC_FILTER = 'ndp','NonExonic','bitgt','gdc_pon'are removed. - Remaining variants with
SOMATIC != nullare included in the Somatic MAF. - Remaining variants with
dbSNP_RS = 'novel'ornullare included in the Somatic MAF. - Remaining variants are removed.
- Variants with
- Removal of the following columns:
- vcf_region
- vcf_info
- vcf_format
- vcf_tumor_gt
- vcf_normal_gt
- GDC_Valid_Somatic
- Set values to be blank in the following columns that may contain information about germline genotypes:
- Match_Norm_Seq_Allele1
- Match_Norm_Seq_Allele2
- Match_Norm_Validation_Allele1
- Match_Norm_Validation_Allele2
- n_ref_count
- n_alt_count
Protected MAF File Structure
The table below describes the columns in a protected MAF and their definitions. Note that the somatic (open-access) MAF structure is the same except for having the last six columns removed.
| Column | Description |
|---|---|
| 1 - Hugo_Symbol | HUGO symbol for the gene (HUGO symbols are always in all caps). "Unknown" is used for regions that do not correspond to a gene |
| 2 - Entrez_Gene_Id | Entrez gene ID (an integer). "0" is used for regions that do not correspond to a gene region or Ensembl ID |
| 3 - Center | One or more genome sequencing center reporting the variant |
| 4 - NCBI_Build | The reference genome used for the alignment (GRCh38) |
| 5 - Chromosome | The affected chromosome (chr1) |
| 6 - Start_Position | Lowest numeric position of the reported variant on the genomic reference sequence. Mutation start coordinate |
| 7 - End_Position | Highest numeric genomic position of the reported variant on the genomic reference sequence. Mutation end coordinate |
| 8 - Strand | Genomic strand of the reported allele. Currently, all variants will report the positive strand: '+' |
| 9 - Variant_Classification | Translational effect of variant allele |
| 10 - Variant_Type | Type of mutation. TNP (tri-nucleotide polymorphism) is analogous to DNP (di-nucleotide polymorphism) but for three consecutive nucleotides. ONP (oligo-nucleotide polymorphism) is analogous to TNP but for consecutive runs of four or more (SNP, DNP, TNP, ONP, INS, DEL, or Consolidated) |
| 11 - Reference_Allele | The plus strand reference allele at this position. Includes the deleted sequence for a deletion or "-" for an insertion |
| 12 - Tumor_Seq_Allele1 | Primary data genotype for tumor sequencing (discovery) allele 1. A "-" symbol for a deletion represents a variant. A "-" symbol for an insertion represents wild-type allele. Novel inserted sequence for insertion does not include flanking reference bases |
| 13 - Tumor_Seq_Allele2 | Tumor sequencing (discovery) allele 2 |
| 14 - dbSNP_RS | The rs-IDs from the dbSNP database, "novel" if not found in any database used, or null if there is no dbSNP record, but it is found in other databases |
| 15 - dbSNP_Val_Status | The dbSNP validation status is reported as a semicolon-separated list of statuses. The union of all rs-IDs is taken when there are multiple |
| 16 - Tumor_Sample_Barcode | Aliquot barcode for the tumor sample |
| 17 - Matched_Norm_Sample_Barcode | Aliquot barcode for the matched normal sample |
| 18 - Match_Norm_Seq_Allele1 | Primary data genotype. Matched normal sequencing allele 1. A "-" symbol for a deletion represents a variant. A "-" symbol for an insertion represents wild-type allele. Novel inserted sequence for insertion does not include flanking reference bases (cleared in somatic MAF) |
| 19 - Match_Norm_Seq_Allele2 | Matched normal sequencing allele 2 |
| 20 - Tumor_Validation_Allele1 | Secondary data from orthogonal technology. Tumor genotyping (validation) for allele 1. A "-" symbol for a deletion represents a variant. A "-" symbol for an insertion represents wild-type allele. Novel inserted sequence for insertion does not include flanking reference bases |
| 21 - Tumor_Validation_Allele2 | Secondary data from orthogonal technology. Tumor genotyping (validation) for allele 2 |
| 22 - Match_Norm_Validation_Allele1 | Secondary data from orthogonal technology. Matched normal genotyping (validation) for allele 1. A "-" symbol for a deletion represents a variant. A "-" symbol for an insertion represents wild-type allele. Novel inserted sequence for insertion does not include flanking reference bases (cleared in somatic MAF) |
| 23 - Match_Norm_Validation_Allele2 | Secondary data from orthogonal technology. Matched normal genotyping (validation) for allele 2 (cleared in somatic MAF) |
| 24 - Verification_Status | Second pass results from independent attempt using same methods as primary data source. Generally reserved for 3730 Sanger Sequencing |
| 25 - Validation_Status | Second pass results from orthogonal technology |
| 26 - Mutation_Status | An assessment of the mutation as somatic, germline, LOH, post transcriptional modification, unknown, or none. The values allowed in this field are constrained by the value in the Validation_Status field |
| 27 - Sequencing_Phase | TCGA sequencing phase (if applicable). Phase should change under any circumstance that the targets under consideration change |
| 28 - Sequence_Source | Molecular assay type used to produce the analytes used for sequencing. Allowed values are a subset of the SRA 1.5 library_strategy field values. This subset matches those used at CGHub |
| 29 - Validation_Method | The assay platforms used for the validation call |
| 30 - Score | Not in use |
| 31 - BAM_File | Not in use |
| 32 - Sequencer | Instrument used to produce primary sequence data |
| 33 - Tumor_Sample_UUID | GDC aliquot UUID for tumor sample |
| 34 - Matched_Norm_Sample_UUID | GDC aliquot UUID for matched normal sample |
| 35 - HGVSc | The coding sequence of the variant in HGVS recommended format |
| 36 - HGVSp | The protein sequence of the variant in HGVS recommended format. "p.=" signifies no change in the protein |
| 37 - HGVSp_Short | Same as the HGVSp column, but using 1-letter amino-acid codes |
| 38 - Transcript_ID | Ensembl ID of the transcript affected by the variant |
| 39 - Exon_Number | The exon number (out of total number) |
| 40 - t_depth | Read depth across this locus in tumor BAM |
| 41 - t_ref_count | Read depth supporting the reference allele in tumor BAM |
| 42 - t_alt_count | Read depth supporting the variant allele in tumor BAM |
| 43 - n_depth | Read depth across this locus in normal BAM |
| 44 - n_ref_count | Read depth supporting the reference allele in normal BAM (cleared in somatic MAF) |
| 45 - n_alt_count | Read depth supporting the variant allele in normal BAM (cleared in somatic MAF) |
| 46 - all_effects | A semicolon delimited list of all possible variant effects, sorted by priority ([Symbol,Consequence,HGVSp_Short,Transcript_ID,RefSeq,HGVSc,Impact,Canonical,Sift,PolyPhen,Strand]) |
| 47 - Allele | The variant allele used to calculate the consequence |
| 48 - Gene | Stable Ensembl ID of affected gene |
| 49 - Feature | Stable Ensembl ID of feature (transcript, regulatory, motif) |
| 50 - Feature_type | Type of feature. Currently one of Transcript, RegulatoryFeature, MotifFeature (or blank) |
| 51 - One_Consequence | The single most severe consequence, based on sequence ontology terms, among all consequence annotations for a variant across transcripts |
| 52 - Consequence | Consequence type of this variant; sequence ontology terms |
| 53 - cDNA_position | Relative position of base pair in the cDNA sequence as a fraction. A "-" symbol is displayed as the numerator if the variant does not appear in cDNA |
| 54 - CDS_position | Relative position of base pair in coding sequence. A "-" symbol is displayed as the numerator if the variant does not appear in coding sequence |
| 55 - Protein_position | Relative position of affected amino acid in protein. A "-" symbol is displayed as the numerator if the variant does not appear in coding sequence |
| 56 - Amino_acids | Only given if the variation affects the protein-coding sequence |
| 57 - Codons | The alternative codons with the variant base in upper case |
| 58 - Existing_variation | Known identifier of existing variation |
| 59 - ALLELE_NUM | Allele number from input; 0 is reference, 1 is first alternate etc. |
| 60 - DISTANCE | Shortest distance from the variant to transcript |
| 61 - TRANSCRIPT_STRAND | The DNA strand (1 or -1) on which the transcript/feature lies |
| 62 - SYMBOL | The gene symbol |
| 63 - SYMBOL_SOURCE | The source of the gene symbol |
| 64 - HGNC_ID | Gene identifier from the HUGO Gene Nomenclature Committee if applicable |
| 65 - BIOTYPE | Biotype of transcript |
| 66 - CANONICAL | A flag (YES) indicating that the VEP-based canonical transcript, the longest translation, was used for this gene. If not, the value is null |
| 67 - CCDS | The CCDS identifier for this transcript, where applicable |
| 68 - ENSP | The Ensembl protein identifier of the affected transcript |
| 69 - SWISSPROT | UniProtKB/Swiss-Prot accession |
| 70 - TREMBL | UniProtKB/TrEMBL identifier of protein product |
| 71 - UNIPARC | UniParc identifier of protein product |
| 72 - RefSeq | RefSeq identifier for this transcript |
| 73 - SIFT | The SIFT prediction and/or score, with both given as prediction (score) |
| 74 - PolyPhen | The PolyPhen prediction and/or score |
| 75 - EXON | The exon number (out of total number) |
| 76 - INTRON | The intron number (out of total number) |
| 77 - DOMAINS | The source and identifier of any overlapping protein domains |
| 78 - GMAF | Non-reference allele and frequency of existing variant in 1000 Genomes |
| 79 - AFR_MAF | Non-reference allele and frequency of existing variant in 1000 Genomes combined African population |
| 80 - AMR_MAF | Non-reference allele and frequency of existing variant in 1000 Genomes combined American population |
| 81 - ASN_MAF | Non-reference allele and frequency of existing variant in 1000 Genomes combined Asian population |
| 82 - EAS_MAF | Non-reference allele and frequency of existing variant in 1000 Genomes combined East Asian population |
| 83 - EUR_MAF | Non-reference allele and frequency of existing variant in 1000 Genomes combined European population |
| 84 - SAS_MAF | Non-reference allele and frequency of existing variant in 1000 Genomes combined South Asian population |
| 85 - AA_MAF | Non-reference allele and frequency of existing variant in NHLBI-ESP African American population |
| 86 - EA_MAF | Non-reference allele and frequency of existing variant in NHLBI-ESP European American population |
| 87 - CLIN_SIG | Clinical significance of variant from dbSNP as annotated in ClinVar |
| 88 - SOMATIC | Somatic status of each ID reported under Existing_variation (0, 1, or null) |
| 89 - PUBMED | Pubmed ID(s) of publications that cite existing variant |
| 90 - MOTIF_NAME | The source and identifier of a transcription factor binding profile aligned at this position |
| 91 - MOTIF_POS | The relative position of the variation in the aligned TFBP |
| 92 - HIGH_INF_POS | A flag indicating if the variant falls in a high information position of a transcription factor binding profile (TFBP) (Y, N, or null) |
| 93 - MOTIF_SCORE_CHANGE | The difference in motif score of the reference and variant sequences for the TFBP |
| 94 - IMPACT | The impact modifier for the consequence type |
| 95 - PICK | Indicates if this block of consequence data was picked by VEP's pick feature (1 or null) |
| 96 - VARIANT_CLASS | Sequence Ontology variant class |
| 97 - TSL | Transcript support level, which is based on independent RNA analyses |
| 98 - HGVS_OFFSET | Indicates by how many bases the HGVS notations for this variant have been shifted |
| 99 - PHENO | Indicates if existing variant is associated with a phenotype, disease or trait (0, 1, or null) |
| 100 - MINIMISED | Alleles in this variant have been converted to minimal representation before consequence calculation (1 or null) |
| 101 - ExAC_AF | Global Allele Frequency from ExAC |
| 102 - ExAC_AF_Adj | Adjusted Global Allele Frequency from ExAC |
| 103 - ExAC_AF_AFR | African/African American Allele Frequency from ExAC |
| 104 - ExAC_AF_AMR | American Allele Frequency from ExAC |
| 105 - ExAC_AF_EAS | East Asian Allele Frequency from ExAC |
| 106 - ExAC_AF_FIN | Finnish Allele Frequency from ExAC |
| 107 - ExAC_AF_NFE | Non-Finnish European Allele Frequency from ExAC |
| 108 - ExAC_AF_OTH | Other Allele Frequency from ExAC |
| 109 - ExAC_AF_SAS | South Asian Allele Frequency from ExAC |
| 110 - GENE_PHENO | Indicates if gene that the variant maps to is associated with a phenotype, disease or trait (0, 1, or null) |
| 111 - FILTER | Copied from input VCF. This includes filters implemented directly by the variant caller and other external software used in the DNA-Seq pipeline. See below for additional details. |
| 112 - CONTEXT | The reference allele per VCF specs, and its five flanking base pairs |
| 113 - src_vcf_id | GDC Submitter_ID for the input VCF file |
| 114 - tumor_bam_uuid | GDC Submitter_ID for the tumor bam file |
| 115 - normal_bam_uuid | GDC Submitter_ID for the normal bam file |
| 116 - case_id | GDC UUID for the case |
| 117 - GDC_FILTER | GDC filters applied universally across all MAFs |
| 118 - COSMIC | Overlapping COSMIC variants |
| 119 - MC3_Overlap | Indicates whether this region overlaps with an MC3 variant for the same sample pair |
| 120 - GDC_Validation_Status | GDC implementation of validation checks. See notes section (#5) below for details |
| 121 - GDC_Valid_Somatic | True or False (not in somatic MAF) |
| 122 - vcf_region | Colon separated string containing the CHROM, POS, ID, REF, and ALT columns from the VCF file (e.g., chrZ:20:rs1234:A:T) (not in somatic MAF) |
| 123 - vcf_info | INFO column from VCF (not in somatic MAF) |
| 124 - vcf_format | FORMAT column from VCF (not in somatic MAF) |
| 125 - vcf_tumor_gt | Tumor sample genotype column from VCF (not in somatic MAF) |
| 126 - vcf_normal_gt | Normal sample genotype column from VCF (not in somatic MAF) |
Notes About GDC MAF Implementation
- Column #4: NCBI_Build is GRCh38 by default
- Column #32: Sequencer includes the sequencers used. If different sequencers were used to generate normal and tumor data, the normal sequencer is listed first.
- Column #61: VEP name "STRAND" is changed to TRANSCRIPT_STRAND to avoid confusion with Column#8 "Strand"
- Column #94: IMPACT categories are defined by the VEP software and do not necessarily reflect the relative biological influence of each mutation.
- Column #122-125: vcf_info, vcf_format, vcf_tumor_gt, and vcf_normal_gt are the corresponding columns from the VCF files. Including them facilitates parsing specific variant information.
- Column #120: GDC_Validation_Status: GDC also collects TCGA validation sequences. It compares these with variants derived from Next-Generation Sequencing data from the same sample and populates the comparison result in "GDC_Validation_Status".
- "Valid", if the alternative allele(s) in the tumor validation sequence is(are) the same as GDC variant call
- "Invalid", if none of the alternative allele(s) in the tumor validation sequence is the same as GDC variant call
- "Inconclusive" if two alternative allele exists, and one matches while the other does not
- "Unknown" if no validation sequence exists
- Column #121: GDC_Valid_Somatic is TRUE if GDC_Validation_Status is "Valid" and the variant is "Somatic" in validation calls. It is FALSE if these criteria are not met
FILTER Value Definitions (column 111)
- oxog : Signifies that this variant was determined to be an OxoG artifact. This was calculated with D-ToxoG.
- bPcr : Signifies that this variant was determined to be an artifact of bias on the PCR template strand. This was calculated with the DKFZ Bias Filter.
- bSeq : Signifies that this variant was determined to be an artifact of bias on the forward/reverse strand. This was also calculated with the DKFZ Bias Filter.
Impact Categories
VEP
- HIGH (H): The variant is assumed to have high (disruptive) impact in the protein, probably causing protein truncation, loss of function, or triggering nonsense mediated decay
- MODERATE (M): A non-disruptive variant that might change protein effectiveness
- LOW (L): Assumed to be mostly harmless or unlikely to change protein behavior
- MODIFIER (MO): Usually non-coding variants or variants affecting non-coding genes, where predictions are difficult or there is no evidence of impact
PolyPhen
- probably damaging (PR): It is with high confidence supposed to affect protein function or structure
- possibly damaging (PO): It is supposed to affect protein function or structure
- benign (BE): Most likely lacking any phenotypic effect
- unknown (UN): When in some rare cases, the lack of data does not allow PolyPhen to make a prediction
SIFT
- tolerated: Not likely to have a phenotypic effect
- tolerated_low_confidence: More likely to have a phenotypic effect than 'tolerated'
- deleterious: Likely to have a phenotypic effect
- deleterious_low_confidence: Less likely to have a phenotypic effect than 'deleterious'